

Abstract
Background: Glucocorticoids (GCs) are common components in chemotherapeutic protocols for acute lymphoblastic leukemia (ALL). A major obstacle in GC therapy, however, is the gradual acquisition of apoptotic resistance in ALL cells repeatedly treated with these hormones. Previous reports indicate that 15-30% of pediatric ALL samples are resistant to GCs, while in refractory childhood ALL, the prevalence of GC resistance is as high as 70%. Identification of specific molecular mechanisms driving resistance to GC and targeting downstream molecules may lead to the development of new therapeutic strategies.
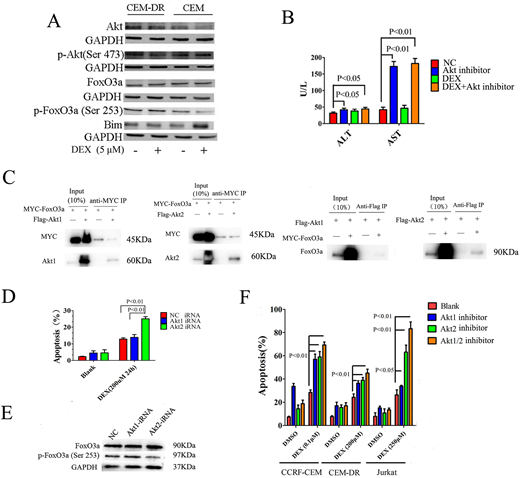
Results: FoxO transcription factor FoxO3a has been shown to regulate apoptosis in lymphocytes. Unphosphorylated FoxO3a can be upregulated by dexamethasone (DEX), which subsequently translocates into the nucleus, upregulates Bim expression and induces apoptosis. In our current study, we cultured a GC-sensitive T-ALL cell line CCRF-CEM with a specific concentration of DEX over multiple passages and obtained a highly resistant cell line, designated CEM-DR. We observed this T-ALL cell line acquires resistance to DEX-mediated killing through abnormal activation of Akt, resulting in inhibition of the FoxO3a/Bim pathway (A). Pharmacologic inhibition of Akt with Akt inhibitor IV effectively restores sensitivity to DEX of CEM-DR by enhancing the FoxO3a/Bim pathway, but shows significant hepatotoxicity with increased serum aspartate aminotransferase (AST) and alanine aminotransferase (ALT) in vivo (B). Common hematologic toxicities and hepatic toxicities with increased AST and ALT of Akt inhibitor have also been reported in the treatment of solid tumors in humans, partially limiting their clinical applicability (Becher OJ et al, Pediatr Blood Cancer 2017). There are two closely related, highly conserved homologues of Akt: Akt-1 and -2, which differ in enzyme function. Our study aimed to investigate the potential role of Akt isoforms Akt1 and Akt2 in the mechanism of GC resistance and explore a more direct and specific target for resistance reversal.
By western blot analysis we observed the expression of Akt2 in intrinsically resistant T-ALL cells Jurkat was significantly higher than that in sensitive CCRF-CEM cells, while Akt1 expression in aforementioned cell lines was similar. The expression of Akt2 also increased synchronously with the increase of half maximal inhibitory concentration (IC50) of DEX in CEM-DR cells, further indicating that Akt2 expression increases in secondarily resistant lymphocytes. A significantly elevated expression of Akt2 not Akt1 were shown in relapsed/refractory ALL patients when compared with the newly diagnosed patients. To detect if Akt2 was able to directly interact with FoxO3a, co-immunoprecipitation assay was employed. MYC-FoxO3a was co-transfected with Flag-Akt1 or Flag-Akt2 into HEK293T cells. The result demonstrated the more presence of Flag-AKT2 than Flag-Akt1 in MYC-FoxO3a immunoprecipitates, suggesting that AKT2 as the major regulator directly interacting with FoxO3a. Reciprocal immunoprecipitation experiments confirmed the closer association between Flag-Akt2 and MYC-FoxO3a (C). Then we used siRNA to down-regulate Akt1 or Akt2 expression in resistant T-ALL cells; we observed GC-induced apoptosis increased significantly (D) after down-regulation of Akt2 expression, along with the expression of p-FoxO3a decreased (E) .
To examine the therapeutic role of Akt isoform specific inhibitors, we treated resistant T-ALL cell with A-674563 (Akt1 inhibitor), CCT128930 (Akt2 inhibitor) or Akti1/2 (Akt1/2 inhibitor). Selective inhibition of Akt2 with CCT128930 more significantly enhances the FoxO3a/Bim signaling pathway, increases DEX-mediated killing of resistant T-ALL cells (F) and effectively reverses GC resistance than Akt1 inhibitor in vitro and in vivo. When exploring the potential influence on viscera by Akt isoform inhibitors in vivo, we found Akt2 inhibitor did not significantly influence hematopoiesis, cardiac and renal functions. Notably, Akt2 inhibitor did not show hepatic toxicities of increased serum ALT, AST and total bilirubin which appear in Akt1 or Akt1/2 inhibition.
Conclusions: Akt2 might serve as a more direct and specific kinase mediating GC resistance through FoxO3a/Bim-signaling pathway in ALL, and targeting Akt2 with CCT128930 may be explored as a promising therapeutic strategy for resistance reversal.
No relevant conflicts of interest to declare.

